
La tecnología CRISPR ha supuesto un cambio de paradigma en el campo de la edición del genoma. En la última década, el número de aplicaciones ha aumentado exponencialmente y los laboratorios de todo el mundo están haciendo uso de esta tecnología para abordar sus hipótesis científicas.
Si quieres saber de dónde viene esta tecnología, cómo funciona, sus aplicaciones y mucho más, ¡echa un vistazo a nuestra nueva publicación en el blog!
1. ¿Qué es CRISPR y cuál es el origen de esta tecnología?
CRISPR son las siglas, en inglés, de `Clustered Interspaced Short Palindromic Repeats´, o lo que es lo mismo, `Repeticiones palindrómicas, cortas, agrupadas y regularmente interespaciadas´. Estas secuencias de ADN repetitivas se descubrieron en bacterias y arqueas. Más tarde se observó que dichas secuencias corresponden a secuencias virales y son la base de un sistema adaptativo de defensa frente a virus que tienen dichos microorganismos.
El mecanismo de este sistema está basado en la incorporación de secuencias cortas del genoma de los virus en el locus CRISPR presente en bacterias y arqueas. De esta forma, tras una infección vírica, estas secuencias se transcriben y permiten destruir el virus. Este mecanismo de defensa también requiere la presencia de genes asociados a CRISPR, abreviados ´Cas´ (del inglés, CRISPR-associated genes), que codifican las proteínas que cortan los ácidos nucleicos del virus. Por este motivo, el sistema se conoce como CRISPR-Cas. Se han descrito diferentes sistemas CRISPR-Cas en bacterias y arqueas, y éstos se diferencian unos de otros tanto en sus componentes como en su mecanismo de acción.
El descubrimiento de este sistema ha resultado en el desarrollo de una gran variedad de herramientas de edición genética que se caracterizan por ser considerablemente más eficientes y fáciles de poner a punto que las tecnologías anteriores.

2. ¿Cómo funciona el sistema CRISPR-Cas9?
El primer sistema CRISPR que se diseñó para editar el genoma de células de mamíferos proviene de Streptococcus Pyogenes. Está constituido por la endonucleasa Cas9, que puede generar una rotura de doble cadena (DSBs, del inglés double strand breaks) tras el reconocimiento de determinada secuencia diana en el ADN. Para guiar a Cas9 al sitio concreto del genoma donde ejercerá su actividad, Cas9 forma un complejo ribonucleoproteico con una secuencia de ARN CRISPR (crARN), que sirve de guía para localizar la secuencia diana, y un par de moléculas de ARN CRISPR trans-activadoras (tracrARNs).
Con el fin de simplificar la aplicación de este sistema como herramienta para la edición genética, se diseñó una única molécula de ARN (sgRNA, del inglés single guide RNA) que aúna las funciones de las moléculas crARN y tracrARN. El sgRNA se puede diseñar a medida para que contenga una secuencia de 20 nucleótidos de longitud que por complementariedad hibridará con la secuencia diana del ADN que se desea modificar.
Es importante destacar que la secuencia de ADN diana debe de estar precedida por una secuencia corta, conocida como PAM (del inglés protospacer adjacent motif) para que pueda cortarse el ADN. En el sistema CRISPR-Cas9, la secuencia PAM (5´ a 3´) es NGG, donde N representa cualquier nucleótido. Otros sistemas CRISPR de otras bacterias se basan en ortólogos de Cas9 que pueden reconocer diferentes secuencias de PAM, ampliando así la elección de las secuencias diana en el ADN que son susceptibles de modificar.
Tras el reconocimiento de la secuencia diana en el ADN por la sgRNA, Cas9 produce la rotura de doble cadena del ADN unos pocos pares de bases corriente arriba de la secuencia PAM. La rotura de doble cadena que se genera desencadenará una respuesta de reparación del ADN que se utilizará para llevar a cabo la modificación genética deseada.
3. ¿Cuáles son las aplicaciones de CRISPR-Cas9 para edición genética?
La rotura de doble cadena generada por las endonucleasas puede ser reparada mediante distintos mecanismos. Dos de los principales mecanismos son los siguientes:
- Unión de extremos no homólogos (NHEJ, del inglés non-homologous end joining): este mecanismo de reparación no es muy preciso y suele cometer errores tras varios ciclos de rotura y reparación. Estos errores pueden consistir en la inserción o deleción de algunos nucleótidos.
- Recombinación homóloga (HDR, del inglés homologous directed recombination): en presencia de un constructo ´donante´ (donor template) el DSB puede repararse sin errores. No obstante, este mecanismo de reparación ocurre con menor eficiencia que el NHEJ ya que requiere que las células se encuentren en fase de división.
En base a los mecanismos de reparación mencionados, se pueden llevar a cabo las siguientes modificaciones genéticas:
- Deleción de un gen (knockout): una estrategia para generar una deleción cosiste en dirigir el gRNA hacia un exón codificante al comienzo del gen de interés. Cuando se produce la unión, Cas corta y el DSB se repara mediante NHEJ, introduciendo así mutaciones que afectan a la pauta de lectura. Como consecuencia, se altera o interrumpe la expresión del gen. Otra estrategia alternativa consiste en utilizar simultáneamente dos gRNAs dirigidas a un mismo gen. De esta forma se podrá eliminar la secuencia presente entre los dos DSBs. Ver Figura 1a.
- Inserciones de genes (knockin): la inserción de una secuencia de ADN en un gen que codifica una proteína se puede lograr proporcionando externamente un constructo ´donante´ que contenga la secuencia de interés. Este constructo se puede incorporar en el genoma mediante recombinación homóloga. Para lograr esto, debe de contener regiones de homología con las secuencias que flanquean al DSB. El constructo puede ser un plásmido de ADN o un oligonucleótido sintético de ADN de hebra simple. Este último se suele emplear en caso de querer introducir una mutación puntual en la región diana. Esta misma estrategia puede utilizarse para introducir marcadores o tags en el genoma de forma precisa para que se expresar de forma endógena. Ver Figura 1b.
- Translocaciones: se pueden originar translocaciones cromosómicas cuando los DSBs se generan en dos cromosomas no homólogos y estos DSB se reparan de forma incorrecta, dando lugar a un intercambio de segmentos entre cromosomas. Mediante el uso de CRISPR-Cas9 se han introducido reordenamientos cromosómicos para imitar, por ejemplo, translocaciones observadas en cáncer o fusiones de genes oncogénicos.
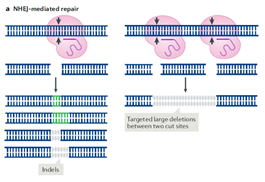
Fuente: (Pickar-Oliver & Gersbach, 2019)
4. ¿Qué es un editor de bases?
Si bien la reparación de DSB mediada por HDR se puede utilizar para introducir mutaciones puntuales, existen algunas limitaciones. En primer lugar, la HDR sólo ocurre durante la división celular por lo que no podemos utilizar esta estrategia en células post-mitóticas. Por otro lado, la introducción de DSB conlleva el riesgo de que se produzcan efectos off-target, es decir, que se produzcan cortes en regiones del genoma con secuencias similares a nuestra secuencia diana.
Una estrategia para solventar estas limitaciones, es el uso de la Cas9 nickase (Cas9n). Se trata de una variante proteica de Cas9 que ha sido diseñada para que únicamente corte una de las hebras del ADN. Esta enzima Cas9 nickase se obtiene mediante desaminación con enzimas de ADN monocatenario, y se conocen como enzimas `editoras de bases´. Se han desarrollado principalmente 2 tipos:
- Cytosine-based editors (CBEs): catalizan la conversión de los pares de bases C/G a T/A.
- Adenine-based editors (ABEs): catalizan la conversión de los pares de bases A/T a G/C.
En ambos casos, la enzima Cas9n (guiada por la gRNA) localiza la enzima desaminasa en la secuencia diana, permitiendo la introducción de un único cambio de nucleótido. La modificación tiene lugar sin generar un DBS.
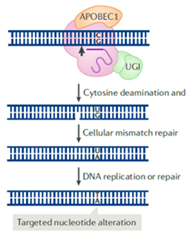
En este ejemplo Cas9 se ha fusionado con APOBEC1, una citidina desaminasa.
Además, se han fusionado dos inhibidores de uracilglicosilasa (UGI) con el editor de base para aumentar la eficiencia de edición de la base.
Reference: (Pickar-Oliver & Gersbach, 2019)
5. ¿Qué hay que tener en cuenta para el diseño de un experimento con CRISPR?
En la última década se han desarrollado numerosas herramientas basadas en CRISPR, que brindan a los investigadores múltiples opciones para editar genes.
A la hora de diseñar un experimento CRISPR, una de las principales cosas a tener en cuenta es el tipo de modificación deseada. Esto determinará cuáles son los reactivos y la estrategia a utilizar. A continuación, describimos brevemente los pasos a tener en cuenta:
- Paso 1: Determinar la modificación genética de interés. Para generar genes knockout el gRNA debe dirigirse al primer exón en 5′ o a un dominio de la proteína que sea esencial. En caso de querer introducir una mutación determinada o knockin, tendremos que estudiar las secuencias PAM cercanas a la mutación para elegir la proteína Cas.
- Paso 2: Seleccionar el sistema de expresión: los componentes de CRISPR-Cas9 se pueden administrar in vivo a través de diferentes sistemas:
- Plásmido: se expresa la proteína Cas9 y el gRNA simultáneamente. Se pueden elegir plásmidos que contengan marcadores o reporters.
- Vector lentiviral: se recomienda su uso para aquellos tipos celulares que sean difíciles de transfectar. Igual que el caso anterior, se puede expresar simultáneamente la proteína Cas9 y el gRNA.
- Vector AVV: puede realizarse la transfección de forma transitoria o estable. Se recomienda utilizar la proteína saCas9, algo más pequeña que Cas9 ya que permite introducir todos los componentes en el vector AAV. Esta estrategia es eficaz para infectar tanto células que se dividan activamente como aquellas que no se dividen.
- Inserción directa de Cas9, mRNA y gRNA: permite la expresión transitoria de los componentes del sistema CRISPR. Se pueden introducir en la célula mediante técnicas de microinyección o electroporación.
- Complejo ribonucleoproteico gRNA/Cas9: La proteína Cas9 se purifica y la molécula de gRNA se transcribe in vitro para una expresión transitoria.
- Paso 3: Seleccionar la secuencia de ADN diana y diseñar el gRNA. Existen bases de datos de gRNAs que han sido validados, así como herramientas de diseño para elegir la mejor guía.
- Paso 4: Para knockins, diseñar el constructo donante. Las construcciones de donantes de HDR pueden administrarse como plásmidos u oligonucleótidos de una sola hebra de ADN (ssDNA, del inglés single stranded DNA). Es importante que el constructo, además de introducir los cambios de interés, introduzca una mutación silenciosa en la secuencia PAM. De esta forma se evitará que el sistema CRISPR vuelva a actuar sobre la nueva secuencia ya incorporada.
- Paso 5: Construir los vectores que contengan la gRNA y la proteína Cas, así como el constructo donante o ssDNA para los knockins.
¿Estás pensando en utilizar la tecnología CRISPR, pero no estás seguro de cómo empezar? En Abyntek te ayudamos a definir la mejor estrategia y a elegir los reactivos necesarios según tus necesidades. ¡Contáctanos para más información!
References:
Anzalone, A. V, Koblan, L. W., & Liu, D. R. (2020). Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nature Biotechnology, 38(7), 824–844. https://doi.org/10.1038/s41587-020-0561-9 https://www.nature.com/articles/s41587-020-0561-9
Pickar-Oliver, A., & Gersbach, C. A. (2019). The next generation of CRISPR-Cas technologies and applications. Nature Reviews. Molecular Cell Biology, 20(8), 490–507. https://doi.org/10.1038/s41580-019-0131-5 https://doi.org/10.1038/s41580-019-0131-5.

